


You are now leaving the aHUSSource.com website.
Alexion is not responsible for content included on third-party websites. Do you want to proceed?

The right diagnosis can help your patients prevent the progression of organ damage and death. Learn how to identify TMA, recognize triggers, and diagnose atypical-HUS.
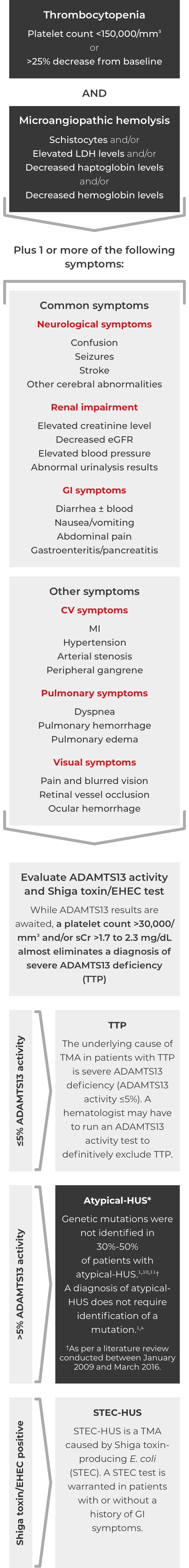
Thrombotic microangiopathy (TMA) manifests as thrombocytopenia, microangiopathic hemolysis, and organ damage. Though TMAs present with similar signs and symptoms, they have distinct underlying causes. A clinical diagnosis of atypical-HUS in a patient with signs of TMA requires exclusion of the other underlying causes.1,4
When you suspect TMA:
Many times, patients experiencing TMA will require urgent specialized care in the ICU. Rapidly identifying atypical-HUS as the underlying cause of TMA is essential for an accurate diagnosis and can help with optimal disease management decisions.1 Review the important factors to consider in pediatric and adult case studies of diagnosing atypical-HUS in an ICU setting.
DOWNLOAD ICU PDF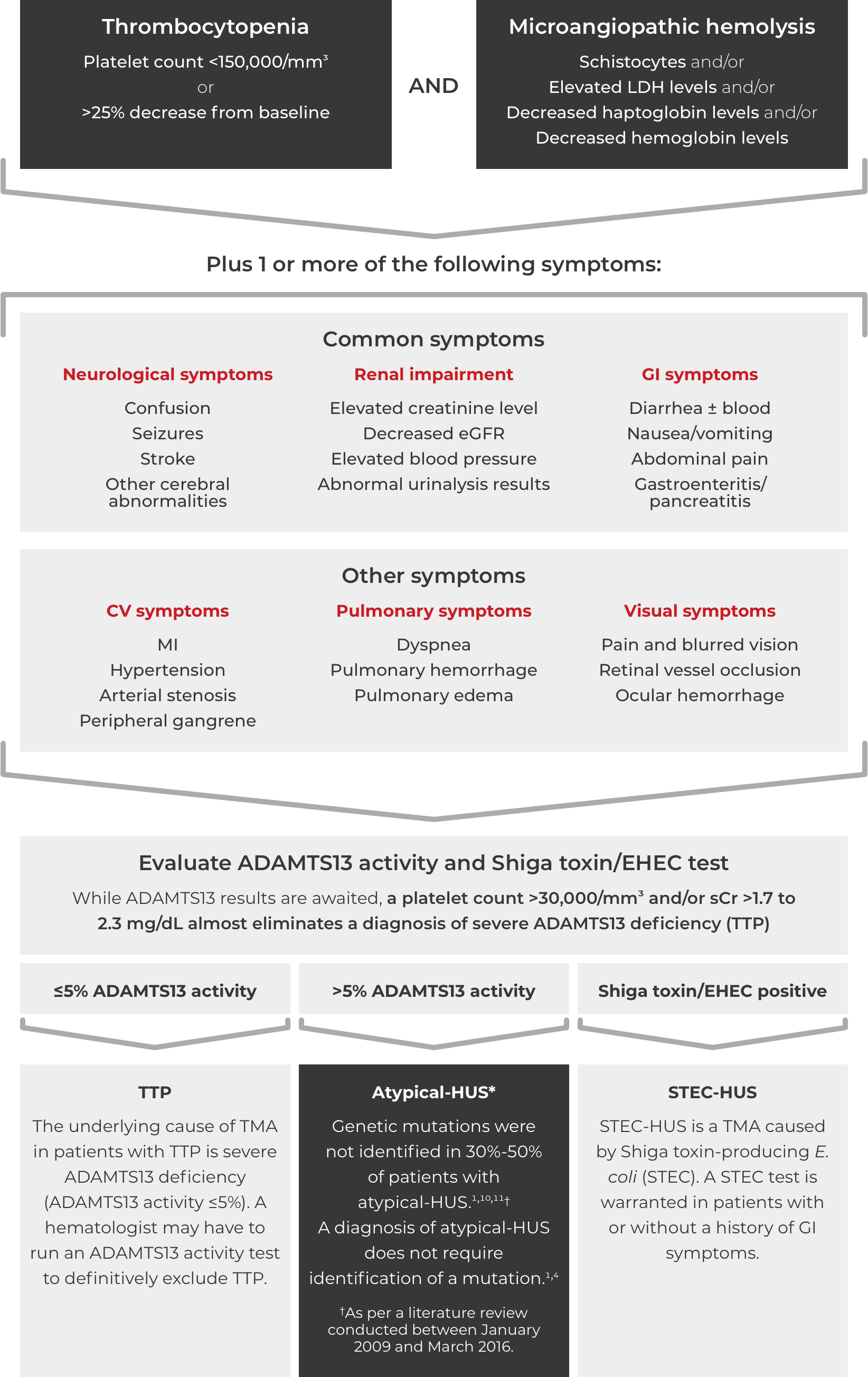
This information is intended as educational information for healthcare providers. It does not replace a healthcare professional’s judgement or clinical diagnosis.
*Where TTP and STEC-HUS have been ruled out, the TMA can be attributed to complement dysregulation. Atypical-HUS is a complement mediated TMA (CM-TMA) that has been more broadly or narrowly defined in different contexts, based on such factors as the presence and nature of a trigger and/or identification of an underlying genetic mutation.4,12,28
ADAMTS13=a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13; CV=cardiovascular; eGFR=estimated glomerular filtration rate; EHEC=enterohemorrhagic Escherichia coli; GI=gastrointestinal; HUS=hemolytic uremic syndrome; LDH=lactate dehydrogenase; MI=myocardial infarction; sCr=serum creatinine; STEC=Shiga toxin-producing Escherichia coli; TTP=thrombotic thrombocytopenic purpura.
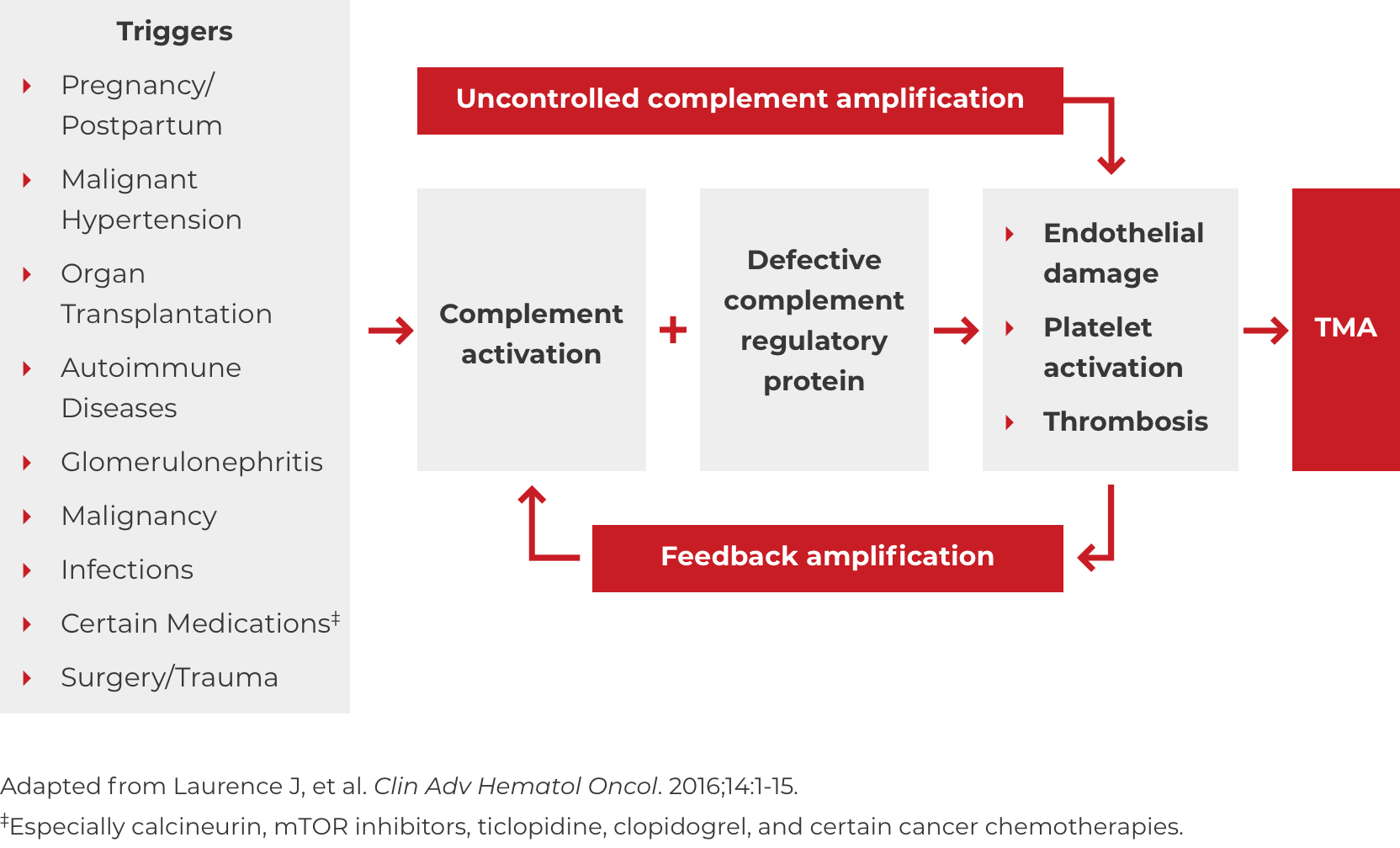
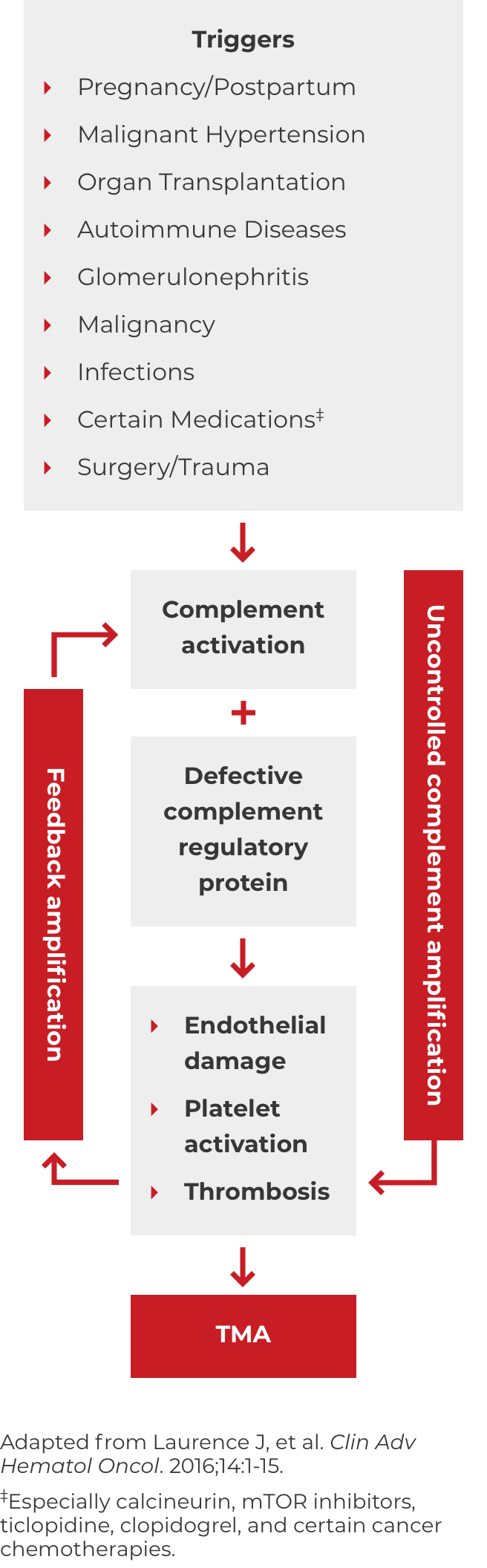
DOWNLOAD DIAGNOSIS TOOL PDFA potential complicating factor in the diagnosis of atypical-HUS is the presence of a triggering condition. There are a number of triggers that can contribute to the onset of atypical-HUS by amplifying complement activation, manifesting with signs and symptoms of TMA.1,2,4 These are some triggers seen in atypical-HUS patients. Additional triggers may exist.
If these signs and symptoms do not rapidly resolve in response to management of the triggering condition, evaluate for atypical-HUS by assessing for STEC-HUS and ADAMTS13 deficiency.4